|
Myeloproliferative neoplasms (MPNs) are clonal hematologic malignancies typified by genetic alterations that hyperactivate JAK-STAT signaling. Chronic MPNs exhibit a propensity for transformation to secondary acute myeloid leukemia (sAML), which carries a dismal prognosis. The overall objective of our work is to better understand the fundamental mechanisms responsible for the initiation and development of MPNs, as well as the factors that drive evolution to sAML. Our laboratory research is broadly organized into two areas:
|
Interrogating Dysregulated Cytokine-Signaling Networks in MPNs
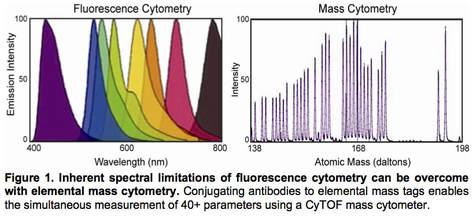
The identification of the JAK2 V617F mutation in MPNs has led to the development of targeted inhibitors of JAK2, which provide significant symptomatic benefit but do not eradicate the underlying malignant clone. These findings suggest that other signaling pathways may be dysregulated in a manner that allows survival of the malignant clone, even when JAK2 is inhibited. We seek to evaluate the role of these altered signaling pathways in MPN development. Mass cytometry is a novel technology that merges aspects of flow cytometry with mass spectrometry and enables the simultaneous measurement of 40+ parameters at the single cell level (Fig 1). This approach enables the characterization of aberrant signaling across the entire hematopoietic continuum. Our efforts with this technology have revealed an extensive network of aberrant signaling in MPN stem/progenitor cells, including the identification of hyperactivated NFkB pathway signaling in MPNs and sAML. Ongoing work is focused on applying mass cytometry to delineate the cellular loci and intracellular signaling pathways that drive abnormal production of inflammatory cytokines (e.g. TNF, IL-6, IL-8) in MPNs.
To complement these efforts, we are utilizing genetic and pharmacologic approaches in the context of mouse models to evaluate the role of these cytokine-signaling pathways in MPN disease development and progression in vivo. Our findings with these approaches have led directly to novel approaches to therapeutic intervention for MPN patients.
The identification of the JAK2 V617F mutation in MPNs has led to the development of targeted inhibitors of JAK2, which provide significant symptomatic benefit but do not eradicate the underlying malignant clone. These findings suggest that other signaling pathways may be dysregulated in a manner that allows survival of the malignant clone, even when JAK2 is inhibited. We seek to evaluate the role of these altered signaling pathways in MPN development. Mass cytometry is a novel technology that merges aspects of flow cytometry with mass spectrometry and enables the simultaneous measurement of 40+ parameters at the single cell level (Fig 1). This approach enables the characterization of aberrant signaling across the entire hematopoietic continuum. Our efforts with this technology have revealed an extensive network of aberrant signaling in MPN stem/progenitor cells, including the identification of hyperactivated NFkB pathway signaling in MPNs and sAML. Ongoing work is focused on applying mass cytometry to delineate the cellular loci and intracellular signaling pathways that drive abnormal production of inflammatory cytokines (e.g. TNF, IL-6, IL-8) in MPNs.
To complement these efforts, we are utilizing genetic and pharmacologic approaches in the context of mouse models to evaluate the role of these cytokine-signaling pathways in MPN disease development and progression in vivo. Our findings with these approaches have led directly to novel approaches to therapeutic intervention for MPN patients.
Genetic Complexity and Clonal Evolution in MPNs
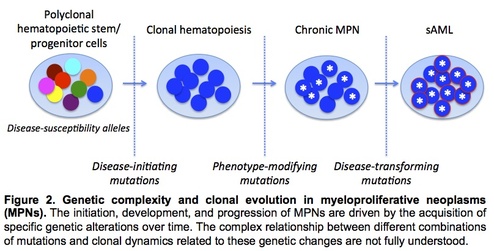
The JAK2 V617F mutation is present in the majority of MPNs, and recent studies have identified recurrent mutations in several other genes including CALR. While the spectrum of recurrent mutations in MPNs has largely been delineated, the nature by which clonal hierarchy specifies MPN clinical phenotype and drives disease evolution remains incompletely understood (Fig 2). We are utilizing next-generation sequencing approaches to unravel this complex interaction between specific mutations and MPN disease phenotype. In a prior study, we utilized whole genome sequencing in conjunction with capture-based deep sequencing validation to recapitulate clonal architecture accompanying disease transformation from a chronic MPN to sAML. In an ongoing project, we are utilizing similar approaches to dissect the genetic basis for distinct clinical phenotypes observed in two subgroups of patients with polycythemia vera. To further uncover how specific mutations interact to modulate MPN disease development, we have generated patient-derived induced pluripotent stem cells (iPSCs). These genetically distinct iPSC clones have been subjected to CRISPR site-directed gene editing to revert JAK2 V617F and/or other driver mutations such that the precise role of these specific mutations (alone and in combination) can be examined to model human MPN pathogenesis.
The JAK2 V617F mutation is present in the majority of MPNs, and recent studies have identified recurrent mutations in several other genes including CALR. While the spectrum of recurrent mutations in MPNs has largely been delineated, the nature by which clonal hierarchy specifies MPN clinical phenotype and drives disease evolution remains incompletely understood (Fig 2). We are utilizing next-generation sequencing approaches to unravel this complex interaction between specific mutations and MPN disease phenotype. In a prior study, we utilized whole genome sequencing in conjunction with capture-based deep sequencing validation to recapitulate clonal architecture accompanying disease transformation from a chronic MPN to sAML. In an ongoing project, we are utilizing similar approaches to dissect the genetic basis for distinct clinical phenotypes observed in two subgroups of patients with polycythemia vera. To further uncover how specific mutations interact to modulate MPN disease development, we have generated patient-derived induced pluripotent stem cells (iPSCs). These genetically distinct iPSC clones have been subjected to CRISPR site-directed gene editing to revert JAK2 V617F and/or other driver mutations such that the precise role of these specific mutations (alone and in combination) can be examined to model human MPN pathogenesis.